
Jenny's Facial Difference
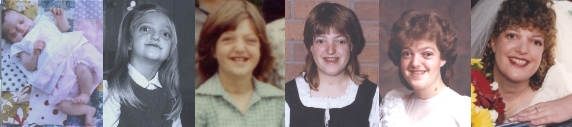
The word 'craniofacial' refers to conditions or syndromes that affect the skull and/or face. Myself, my daughter and my son have this syndrome.
Crouzon syndrome (also known as Craniofacial Dysostosis) was originally described in 1912 by O. Crouzon in Paris in a mother and her daughter. It is caused by a mutation in the FGFR2 (fibroblast growth factor receptor-2) gene or in the FGFR3 gene (known as acanthosis nigricans when a dark discolouration occurs with rough skin in the armpits and groin). These genes were first reported in 1994 by Reardon. Basically this means that when the baby's face and skull are forming in the womb, the cells which form them are over active, so grow too quickly. This means that the bones reach their growth potential too soon, so are stunted. Characteristics of a person with Crouzons are described below. Though like in any condition, symptoms can range from mild to severe, and not all people display all characteristics. Also symptoms can be different in each generation. And Jenny has found from talking to other people who have Crouzons, or their children do, there are also other characteristics that have not been documented.
The main symptoms are:
|
| Facial features - large bulging eyes and a flat face (due to a recessed mid-face) |
Craniosynostosis
Before birth and early in infancy, a child's brain grows very rapidly, reaching 70% of its eventual adult size in the first year. To keep up with this growth, the head must expand rapidly while keeping the brain protected. This is able to happen because a baby's skull bone is made up of a collection of many smaller bones. Where these bones meet are called sutures. Each of these sutures has a name (see the diagram). As the brain grows the sutures expand allowing for rapid symmetrical expansion. In babies with Crouzon syndrome these sutures 'close' or fuse together which means these bones cannot expand for brain growth. If there are sutures which are not fused then that is where the brain will grow towards leading to abnormal head shapes. Because the brain is growing but the skull does not in most cases expand adequately enough for the brain's size, pressure is also put on the brain which if not eased, will lead to mental retardation and eventually death. The brain can also put pressure on the optic nerves at the back of the eye which can lead to blindness.

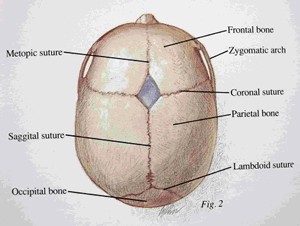
Which sutures are affected varies widely. In typical cases of Crouzons, the coronal sutures which run along the top of the head from ear to ear, are affected making the head high and thin, like in the photo below.
In Jenny's family's case their fusions occurred differently to this. All of Jenny's sutures at birth were fused. In Melissa's case, the coronal, metopic and lambdoid sutures were (front and back). Melissa's sagittal suture on the top of her head was huge to compensate and her brain grew out through this. In Nicholas's case at birth his metopic suture was fused and there was a ridge down the middle of his forehead. He had a very small soft spot, about 1cm round on the top of his head. Nicholas also had reflux which went undiagnosed for 3 months which kept his head turned to the left. This left a flat spot on the left-hand side of the back of his head which over time became less pronounced. By 9 months Nick's lamboid sutures had closed on the back of his head which also caused a flat spot but this was deemed as not being serious enough to need surgery.
|
Jenn |
Melissa |
Nick |



Because of the range of severity in Crouzon syndrome, she children do not require any surgery on their skulls. The brain is able to grow at a normal rate and be accommodated within the skull. In other cases, surgery is very necessary to ensure that intellectual impairment and death, do not occur. In past generations there were babies who died from Crouzon syndrome. (Jenny has heard from nurses that in her own generation there were babies who died so she feels very fortunate that there was a surgeon willing to separate her skull.)
Facial Features
People with Crouzons look similar and could often pass as brothers and sisters. The feature that stands out the most when looking at a person who has Crouzons, are their prominent eyes. These can be of varying degrees, depending on the degree of severity. The bulgy eyes are due to shallow eye orbits (sockets). Along with the big eyes comes a flattened face which makes the eyes look bigger. The face looks flat because the middle of the face - under the eyes down to the top jaw, stop growing prematurely. This leads to the eyes looking more and more prominent and the bottom jaw getting more and more prominent over time.
When Melissa was born she did not look like she had Crouzons. Her eyes were not prominent. But over time with her mid face not growing her eyes have become very noticeable. See below. Jenny and Nicholas both had prominent eyes at birth.
|
Two weeks old |
Three years old |


Other Features Associated with the Syndrome:
Other common features of the syndrome include:
| low set ears | |
| hearing loss | |
| eye sight issues | |
| droopy eyes | |
| beaked nose | |
| sleep apnea due to small nasal passages | |
| high-arched palate | |
| narrow upper jaw | |
| crowded teeth | |
| short upper lip | |
| some cleft lips | |
| chiari in the spine | |
| headaches | |
| seizures | |
| hydrocephalus | |
| kidney problems | |
| heart problems | |
| large broad thumbs and big toes | |
| large hands and feet | |
| gaps between the big toe and the second toe | |
| some have mild to moderate intellectual impairment | |
| and some need permanent tracheas. |
Who Looks after these Children?
All over the world there are clinics set up in the major hospitals. The clinics consist of a multi-disciplinary team of doctors. Usually a team consists of a plastic surgeon, neurosurgeon, geneticist, speech pathologist, orthodontist, audiologist and ophthalmologist. Other specialists may also be involved depending on the symptoms.
The severity of the syndrome will determine what and how many surgeries are required. Generally a cranio vault remodel (cvr) and/or a fronto-orbital advancement (foa) are required in the early years to expand the skull. If hydrocephalus is present a shunt is normally inserted. Some children have a chiari and sphinx in their spine, and require decompression surgery. For the flat mid-face a Lefort III is performed. This can consist of external or internal distraction, or a monoblock. Other surgeries consist of tonsils/adenoids, eye muscle surgery for strabismus, grommets, jaw surgery (Lefort I or II). Most children also have orthodontics to expand the jaws.
Genetics
Crouzon syndrome can 'just happen' or it can be congenital. In Jenny's case there is no family history of the syndrome so it is said to have 'just happened' (medically called a 'sporadic or spontaneous mutation' and is said to occur in 25% - 50% of cases). Jenny has been told that the medical fraternity do not know why the gene mutates, the only factor they have currently identified is the father's age. The gene change though is present in only one of the approximately 24,000 genes which a person has.
Now that Jenny has the syndrome, she has the gene (it is now called 'familial'). The three Woolseys had genetic testing done which confirmed the syndrome. The gene identified in each of them is the FGFR2 (fibroblast growth factor receptor-2) exon 7 cDNA 833G->T (cys278phe). This put in a simple way means that in the FGFR2 gene, the amino acid in the protein at position 278 changed from a cystine which it should have been, to a phenylalanine. This simple change, or mistake, in the type of amino acid caused the face to not grow normally.
The chance of Jenny passing on the gene to each of her children was 50/50 per pregnancy. Melissa received the gene so she has the syndrome and she too has a 50/50 chance of passing the gene on to each of her children and Nicholas received the gene so he has the syndrome and he too has a 50/50 chance of passing the gene on to each of his children. Boys and girls are affected equally. If a child is born without the syndrome then they have the same chance of passing on the gene as the general population. The syndrome does not skip generations but can be so mild that no one realises that someone has it. The incidence is inconsistently reported but seems to range from 1 in 25,000 to 1 in 100,000 (when Jenny was young she was told it was 1 in 250,000)..
